La condensazione aldolica è una reazione comune ed estremamente interessante in chimica organica e, sebbene essa venga comunemente rappresentata come condensazione tra un enolato ed un'aldeide, offre diversi spunti di discussione:


Generalmente dall'enolato Z si ottiene l'allodio syn, mentre da quello E si ottiene anti.



Dieckmann
- Enolato termodinamico e cinetico;
- Isomeria geometrica dell'enolato;
- Diasteroselettività dell'attacco;
- Condensazione con aldeide chirale.
Enolato termodinamico o cinetico?
A seconda delle condizioni di deprotonazione si può cambiare il decorso della reazione: prendiamo in considerazione l'esempio del 2-metilcicloesan-1-one.

É risaputo che l'enolato termodinamico è quello più sostituito, pertanto la reazione 1 porta all'enolato termodinamico, mentre la reazione 2 a quello cinetico. Se l'enolato termodinamico deriva dalla reazione 1, significa che per tale reazione si prediligerà una base che lavori in condizioni di equilibrio (una base debole o <1eq di base forte); per ottenere l'enolato cinetico occorreranno invece condizioni drastiche che non diano tempo al sistema di arrivare all'equilibrio termodinamico (1eq di base forte).

In termini di energia libera di Gibbs, l'enolato termodinamico ha un'energia minore dell'enolato cinetico, ma la barriera di attivazione nel secondo caso è minore. In ogni caso, l'enolato ha un'energia maggiore del chetone reagente.
Isomeria geometrica dell'enolato
Quando viene generato un enolato, esso può essere in forma E o Z:


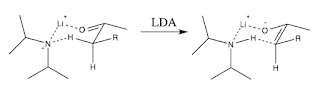
L'enolato che si forma in condizioni cinetiche è generalmente E e la motivazione venne data nel 1976 da Ireland:

Lo stato di transizione per la condensazione alcolica è supposto essere un TS di Zimmermann-Traxler, ossia uno stato di transizione "tight" (stretto) a sedia. Nell'immagine sopra si vede come la presenza dei due gruppi isopropilici dell'ammide costringe il gruppo "R" a disporsi in posizione equatoriale: l'enolo che si forma in prevalenza è l'E.
Se lo stato di transizione viene allargato, ad esempio tramite l'introduzione di HMPA,


il rapporto E/Z diminuisce poiché il fattore sterico risulta essere meno importante.
Diastereoselettività dell'attacco
A seconda dell'enolato che viene generato (E o Z), si ottiene l'aldolo syn o anti.

Generalmente dall'enolato Z si ottiene l'allodio syn, mentre da quello E si ottiene anti.
Condensazione con aldeide chirale
Molto interessante è il caso in cui l'aldeide reagente presenti già un centro stereogenico: in questo caso le due facce dell'aldeide saranno diastereotopiche e le probabilità di attacco delle due facce saranno diverse. In particolare si suppone un TS aperto, in cui il C in alfa al C aldeidico abbia un sostituente S, M ed L in base alla loro taglia e si adopera la proiezione di Newman.

Il sostituente L occupa sempre una faccia diastereotopica, mentre S ed M sono dalla stessa parte; il Nu (ossia l'enolato) attacca dalla parte del gruppo S con angolo di attacco di 107° (Bürgi-Dunitz). Il gruppo L può essere o un gruppo stericamente più grande oppure un alogeno.
Esempi
Per concludere questo post si possono citare alcuni esempi famosi di condensazione aldolica: condensazione di Claisen, Dieckmann, Mukayama, Knoevenagel.Claisen


Dieckmann
 |
| Da Wikipedia |
Mukaiyama
 |
| Da Wikipedia |

0suppvoflec_ka1986 Andy Smith https://wakelet.com/wake/22Xjar9ZuDPRkpOK2VJ6y
RispondiEliminabelviatrilaf
ircae0mic_ze Nathan Albright Avast Premier
RispondiEliminaWonderShare Recoverit
Bandicam
profdecsystro