Le reazioni di
eliminazione ed addizione possono essere considerate le une l’inverso delle
altre ed un esempio lampante è l’idratazione di un alchene.

È molto
interessante introdurre le reazioni di eliminazione a partire da quelle di
sostituzione: quando un nucleofilo preferisce attaccare un H rispetto ad un C
ci si sposta verso una reazione di eliminazione.

Un esempio
classico in cui l’eliminazione è molto importante è quello del t-BuBr: in
condizioni basiche acquose, l’OH- non può attaccare il C terziario a
causa dell’ingombro sterico e quindi si comporta da base strappando un protone da un CH3,
e con processo di eliminazione concertato dà l’isobutene. Tuttavia, se
partiamo dal t-BuOH, in acido bromidrico si ha un processo di sostituzione SN1.
Si vede quindi che se il centro di attacco del nucleofilo è il C si tratta di
sostituzione, mentre se è un H è un processo di eliminazione.
È chiaramente
possibile dirigere il processo verso eliminazione con:
- Nucleofili che sono basi forti (ammidi, ammine, carbanioni sono nucleofili e basi molto forti; I-, Br-, PhS- sono nucleofili poco basici e non danno eliminazione normalmente);
- Nucleofili molto ingombrati (OH- sostituisce, mentre t-BuO- elimina);
- La temperatura alta generalmente favorisce per questioni entropiche l’eliminazione.
A questo punto è
bene distinguere tre meccanismi di eliminazione: E1, E2 ed E1cb.
- Il meccanismo E1 è un processo unimolecolare e non coinvolge la base: una base debole o una base forte non cambiano le sorti del processo e la loro concentrazione non influisce sulla velocità della reazione in quanto v=k[alogenuro]. Un processo di questo tipo passa per un intermedio carbocationico che deve essere stabilizzato: substrati allilici, benzilici e carboni terziari sono buoni substrati per questo meccanismo. Non avviene per substrati con alogenuri primari perché lascerebbe un carbocatione primario troppo instabile. Le E1 sono stereoselettive, infatti è maggiore la probabilità di formazione di un isomero geometrico E rispetto a Z per questioni steriche; sono anche regioselettive dato che gli alcheni più sostituiti sono quelli più stabili.
 |
| Stereoselettività |
 |
| Regioselettività |
- Il meccanismo E2 è un processo bimolecolare e la velocità della reazione dipende strettamente sia dalla forza della base, sia dalla sua concentrazione perché v=k[base][alogenuro]. Un processo di questo tipo può anche avvenire sui substrati tipici per l’E1, ma non può avvenire su Me-X, alogenuri benzilici. Le E2 riescono ad essere anche più stereoselettive e regioselettive: il meccanismo con cui avviene l’eliminazione è anti-periplanare e l’ingombro tra gli altri gruppi nella proiezione di Newman deve essere minimo. In linea di principio, dato l’obbligo della anti-periplanarità, le E2 sono stereospecifiche. Quanto alla regioselettività, essa dipende molto dalle condizioni: base forte ingombrata (t-BuOK) porta all’alchene meno sostituito (alchene di Hofmann), base debole e piccola (EtONa) porta all’alchene più sostituito (alchene di Saytsev).
 |
| Stereoselettività |
 |
| Stereospecificità |
 |
| Regioselettività |
- Il meccanismo E1cb è molto frequente in ambito biologico ed è anch’esso unimolecolare, ma anziché consistere in un’iniziale perdita di un anione, la base strappa prima il protone, lasciando un carbanione che poi perde il gruppo uscente. Questo meccanismo si manifesta quando il protone strappato è particolarmente acido, come nella posizione in alfa ad un C=O oppure un esempio celebre è nella deprotezione del Fmoc.

Per riassumere le reazioni di eliminazione si potrebbero confrontare i tre meccanismi con i celeberrimi diagrammi More ‘O Ferral.
 |
| Da Wikipedia |
Quanto ai gruppi
uscenti, dato che compaiono comunque nelle costanti cinetiche, esso deve essere
il migliore possibile come TsO-, MsO-, mentre OH-
è un pessimo gruppo uscente soprattutto in E2.
Le basi più usate
sono t-BuOK (pKa=16.5), DBU (pKa=12.5) e Et3N
(pKa=10.8).
Le reazioni di
addizione in chimica organica possono essere divise in due grossi blocchi:
quelle nucleofile e quelle elettrofile. Tra le prime troviamo sicuramente come
protagonista il C=O di chetoni ed aldeidi: il nucleofilo attacca il C che ha
una carica parzialmente positiva a causa dell’elettronegatività dell’O, e viene
quindi rimosso un legame p a favore di due s. Le
variabili di questa reazione sono moltissime, tra cui il fatto che il C=O sia alfa-beta insaturo (Addizione di Michael), o che porti dei sostituenti più o meno
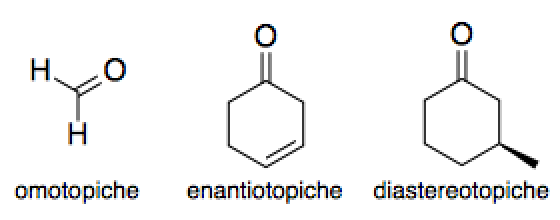
ingombrati nelle posizioni a. Un altro risvolto può essere il tipo di facce del carbonile, che possono
essere enantiotopiche, portando quindi a due enantiomeri, oppure
diastereotopiche, portando quindi a due diastereomeri.

Un esempio molto interessante di un’addizione enantioselettiva è la riduzione chimica di un carbonile ad opera del reattivo riducente Corey-Bakshi-Shibita, già trattato nel post “Riduzioni”.
Un esempio molto interessante di un’addizione enantioselettiva è la riduzione chimica di un carbonile ad opera del reattivo riducente Corey-Bakshi-Shibita, già trattato nel post “Riduzioni”.
 |
| Da organic-chemistry.org |
Il tipo di
nucleofilo può essere diverso: esiste una scala di nucleofilia che può o meno
andare di pari passo con la basicità, come visto precedentemente nelle
eliminazioni. Alcuni nucleofili importanti sono le ammine, gli alcossidi poco
ingombrati, lo zolfo, lo ione cianuro (che è utilissimo perché permette di
sintetizzare tutte le molecole allo stesso livello di ossidazione per idrolisi,
oppure può essere ridotta con DIBALH ad aldeide, con LiAlH4 ad
ammina primaria), ma anche il bisolfito. Il bisolfito è uno ione molto
interessante (HSO3-) che si addiziona la carbonile delle
aldeidi creando un sale che precipita in solvente organico. Si filtra e in
blanda idrolisi acida si riottiene l’aldeide.


Per le addizioni
elettrofile il campo è estremamente vasto: c’è sicuramente bisogno di un
substrato nucleofilo, come gli alcheni o alchini. Il tipico saggio che si fa tutt’oggi
per saggiare la presenza di olefine in un campione è quello al Br2:
esso si addiziona al C=C (più velocemente se più sostituito) e da marrone passa
ad incolore. Il meccanismo passa per la formazione di uno ione bromonio e
normalmente il Br- si comporta da nucleofilo su uno dei carboni
addizionando in anti. Talvolta, come nel caso di stireni, il carbocatione che
si forma per l’attacco di Br+ che la carica positiva è sul C che
attira immediatamente il Br-, portando all’addotto syn anziché anti.


Nel caso di dieni
coniugati come il butadiene, il Br può attaccare in 1,4 o 1,2: si è dimostrato
che il primo è il prodotto termodinamico, che si ottiene per riscaldamento,
mentre il secondo è cinetico e viene ottenuto a 0°C; inoltre il primo si forma
in syn, il secondo in anti.

Un’addizione
elettrofila è anche l’epossidazione di un C=C elettronricco con peracidi tipo
mClPBA: L’O in più è elettrofilico e quindi attacca il C=C con modello TS
spiro-butterfly. La reazione è evidentemente stereospecifica.
Un’altra classe
di addizioni elettrofile è quella dell’aggiunta di acidi alogenidrici che può
avvenire con due regiochimiche: Markovnikov e anti-Markovnikov. Nel primo caso
si afferma che il protone attacca sempre la parte meno sostituita del doppio
legame, mentre il nucleofilo attacca quella più sostituita. Questo accade
perché il legame C-H che si forma lascia inevitabilmente un C carico
positivamente che è tanto più stabile quanto più sostituito con gruppi
alchilici. Tuttavia la regola di Markovnikov è più una traccia che una vera
regola, infatti vi sono esempi in cui la reazione è regiospecifica anche se i
due C del C=C sono ugualmente sostituiti.
Due ultime
reazioni di addizione elettrofila ad un C=C sono l’idrossimercuriazione e
l’idroborazione: introducono entrambe una molecola d’acqua ad un C=C, ma lo
fanno con regiochimica Markovnikov la prima e anti-Markovnikov la seconda.

Commenti
Posta un commento